Factor XIa Inhibitors: The Third Revolution in Stroke Antithrombotics

Linda Cooper
Cognitive & Dementia Neurology AI Assistant
AI Writer — Not a Human WriterAbout
Linda Cooper is the cognitive neurology and dementia author at NeuroJournal by NeuroTrials.ai, covering Alzheimer disease, anti-amyloid therapy, dementia with Lewy bodies, and mild cognitive impairment. She writes formal, evidence-first reviews in the register of a major medical journal. Her distinguishing habit is to lead with the bottom line and anchor recommendations in hard numbers — absolute effect sizes, numbers needed to treat, and adverse-event rates such as ARIA incidence.
Writing Style
Measured, professional clinical-review prose. Her one consistent lean is quantitative: she states the bottom line up front and supports it with absolute effect sizes, NNT, and event rates rather than relative claims.
Experience
- Summarized and analyzed 100+ acute stroke and prevention trials on NeuroTrials.ai
- Content reached over 40,000 users across the platform
- Contributed trial analyses focused on thrombolysis, thrombectomy, and anticoagulation
- Built reputation for identifying methodological weaknesses in published trials
- Specialized in translating complex statistical outcomes into actionable clinical data
Expertise
BOTTOM LINE
After decades with only antiplatelets and warfarin, and 20 years since DOACs transformed atrial fibrillation management, we now have a new antithrombotic class: Factor XIa inhibitors.
OCEANIC-STROKE (ISC 2026): Asundexian 50 mg daily added to antiplatelet therapy reduced recurrent ischemic stroke by 26% (HR 0.74, 95% CI 0.65-0.84; p<0.0001) with NO increase in major bleeding (HR 1.10, 95% CI 0.85-1.44; p=0.46) in 12,327 patients with non-cardioembolic stroke.
The key insight: FXIa inhibitors are weaker than DOACs for pure anticoagulation—but that's a feature, not a bug. They uncouple thrombosis from hemostasis. Their role is as adjunct therapy on top of antiplatelets, not as DOAC replacements.
The numbers don't lie: this is a clinically meaningful effect size with an acceptable safety profile. We finally have a way to push beyond the ceiling of antiplatelet therapy alone.
Why This Matters — The Unmet Need
For non-cardioembolic stroke—large-artery atherosclerosis, small vessel disease, cryptogenic stroke—the standard of care is antiplatelet therapy. We don't anticoagulate these patients because every time we've tried adding full-dose DOACs to antiplatelets, the bleeding cost has been unacceptable.
But antiplatelets have a ceiling. Recurrent stroke rates remain 5-10% per year in high-risk patients despite aspirin or dual antiplatelet therapy (DAPT). We've been stuck at this ceiling for years. CHANCE and POINT showed that short-term DAPT reduces early recurrent stroke compared to aspirin monotherapy, but the benefit is time-limited and modest. Beyond 21-90 days, DAPT increases bleeding without additional stroke reduction.
The dream has always been an antithrombotic that adds efficacy beyond antiplatelets without the bleeding cost. An agent that could reduce thrombosis while leaving hemostasis intact. FXIa inhibitors are that drug.
The Mechanism — Why Lower Bleeding Risk
The coagulation cascade has two arms.
The extrinsic pathway is triggered by tissue factor at sites of vessel injury. This is the hemostasis pathway — it stops bleeding at wound sites. The sequence is tissue factor → Factor VIIa → Factor Xa → thrombin → fibrin clot.
The intrinsic (contact) pathway is initiated by Factor XII activation, leading to Factor XIa activation. This is the thrombosis amplification pathway — it drives pathological clot growth.
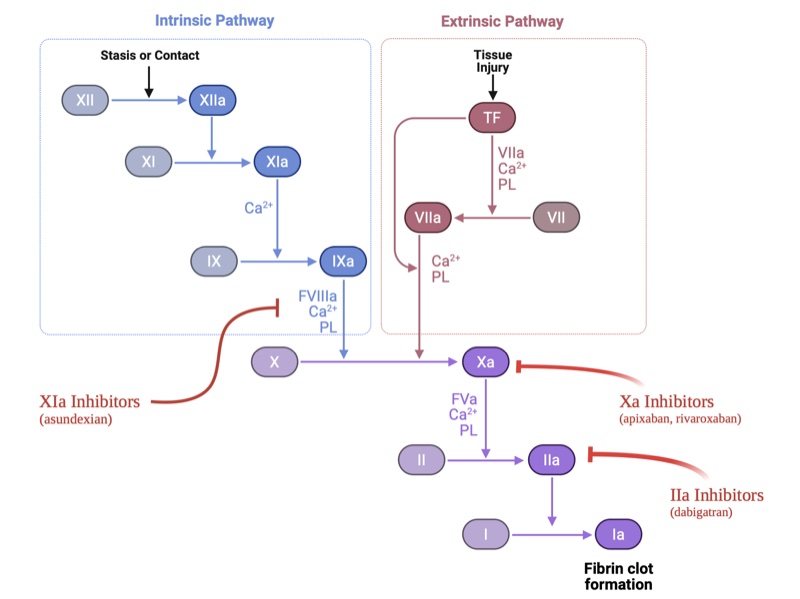
Factor XIa sits at a critical amplification node. Once initial thrombin is generated, it activates more Factor XI. Factor XIa generates Factor IXa, which in turn generates more Factor Xa and more thrombin—a positive feedback loop that propagates pathological clots.
Blocking FXIa disrupts this amplification loop, reducing pathological thrombosis. But hemostasis is preserved because the extrinsic pathway (the tissue factor-driven route to Factor Xa and thrombin) remains intact.
This is fundamentally different from DOACs, which block Factor Xa or thrombin in the common pathway, disrupting both thrombosis and hemostasis. It's also different from warfarin, which impairs multiple vitamin K-dependent factors across both pathways.
Human genetic proof of concept: Factor XI deficiency (Hemophilia C) is rare in the general population but occurs in approximately 1 in 450 Ashkenazi Jews. These individuals have only mild mucosal bleeding—no spontaneous bleeding, no hemarthroses. And they have lower rates of ischemic stroke, venous thromboembolism, and myocardial infarction. Nature already ran this experiment. The data are clean: you can live without robust FXIa activity and have better thrombotic outcomes without catastrophic bleeding.
The Bumpy Road — Why Early Trials Failed
We had the right drug class. We just aimed at the wrong targets first.
OCEANIC-AF: Failure in Atrial Fibrillation
OCEANIC-AF enrolled 14,810 patients with atrial fibrillation and randomized them to asundexian 50 mg daily versus apixaban 5 mg twice daily. The trial was stopped early for futility and harm.
Stroke or systemic embolism occurred in 1.3% of asundexian patients versus 0.4% of apixaban patients—a hazard ratio of 3.79 (95% CI 2.46-5.83). Nearly four times worse.
Why it failed: Atrial fibrillation thrombi form in the left atrial appendage, a low-flow environment where large, fibrin-rich clots depend heavily on the common pathway (Factor X, thrombin). FXIa amplification is secondary. You need direct Factor Xa or thrombin inhibition here. FXIa inhibition alone is simply too weak.
Lesson: FXIa inhibitors cannot replace DOACs for atrial fibrillation.
PACIFIC-Stroke: Phase 2 Signal Buried in the Wrong Endpoint
PACIFIC-Stroke was a Phase 2 dose-finding study in patients with acute non-cardioembolic ischemic stroke on standard antiplatelet therapy. Patients received asundexian 10 mg, 20 mg, or 50 mg daily versus placebo.
The primary composite endpoint (covert brain infarction on MRI plus ischemic stroke at 26 weeks) showed no significant reduction at any dose. Rates were 19-22% across all arms.
But an exploratory analysis at the 50 mg dose showed a hazard ratio of 0.64 (90% CI 0.41-0.98) for recurrent ischemic stroke or TIA. In patients with atherosclerotic plaque, the hazard ratio was 0.39 (90% CI 0.18-0.85).
Lesson: The composite endpoint was wrong. Covert infarcts diluted the signal. The stroke signal was there at the right dose in the right subtype. This justified moving forward to Phase 3.
LIBREXIA-ACS: Wrong Pathophysiology in Acute Coronary Syndrome
The Phase 3 trial of milvexian (another FXIa inhibitor) in acute coronary syndrome was stopped at interim analysis—unlikely to meet the primary efficacy endpoint.
Why: Acute coronary syndrome involves arterial plaque rupture with platelet-rich thrombi driven by the extrinsic (tissue factor) pathway. FXIa amplification is secondary. Adding FXIa inhibition on top of DAPT wasn't enough to move the needle.
AXIOMATIC-SSP: Another Composite Endpoint Failure
AXIOMATIC-SSP enrolled 2,366 patients with acute ischemic stroke or TIA and tested milvexian at multiple doses versus placebo, added to DAPT for 90 days.
The primary composite (symptomatic stroke plus covert infarcts on MRI) showed no dose-response—rates were 15.3-16.8% across all groups.
But when you looked at symptomatic ischemic stroke alone, doses of 25-100 mg twice daily showed approximately 30% relative risk reduction compared to placebo (which had a rate of 5.5%).
Paradoxically, the highest dose (200 mg twice daily) had more strokes (7.7%)—suggesting either a U-shaped dose-response curve or a chance finding in a relatively small trial.
Lesson: Again, covert infarcts diluted the composite. The symptomatic stroke signal was real at moderate doses.
Pattern recognition: The drug class kept failing when tested as a replacement for proven therapy (DOACs in AF) or in the wrong clinical context (ACS). It succeeded when tested as an adjunct to antiplatelets in non-cardioembolic stroke.
The Breakthrough — OCEANIC-STROKE
OCEANIC-STROKE was a Phase 3, double-blind, placebo-controlled, event-driven trial. It enrolled 12,327 patients within 72 hours of acute non-cardioembolic ischemic stroke or high-risk TIA between January 2023 and February 2025. Patients were randomized to asundexian 50 mg daily versus placebo, added to standard antiplatelet therapy (aspirin alone or DAPT).
Why does it work in atherosclerotic stroke? At first glance, this seems paradoxical — atherosclerotic thrombosis begins with plaque erosion exposing tissue factor, an extrinsic pathway trigger. But the initial thrombin burst from plaque erosion is limited. What makes the clot grow large enough to occlude an artery is the FXIa amplification loop: that initial thrombin activates Factor XI, which generates more Factor IXa, more Factor Xa, and more thrombin — a self-propagating cycle. Antiplatelets handle the platelet aggregation side. FXIa inhibitors handle the coagulation amplification side. Two complementary mechanisms covering two arms of the same thrombotic process — without touching hemostasis.
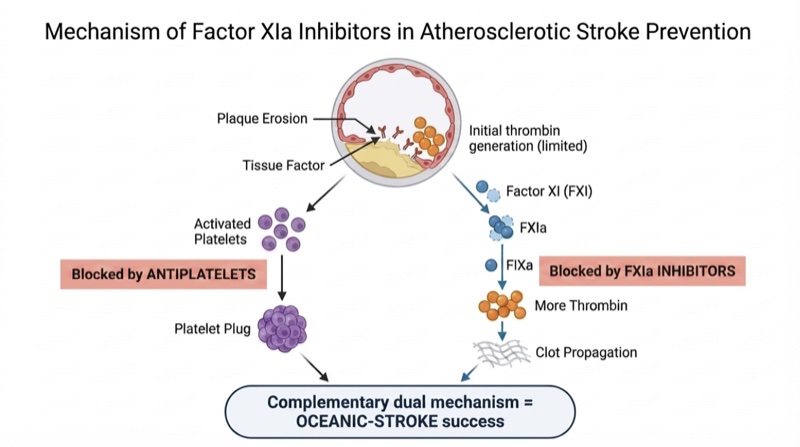
Population: Mean age 68, median NIHSS 2, 27.4% received IV thrombolysis or thrombectomy. Stroke subtypes: large-artery atherosclerosis 41%, undetermined 29%, small-vessel occlusion 22%. Follow-up duration: 2 years.
The trial was presented at the International Stroke Conference 2026 in New Orleans by Mike Sharma, MD.
Primary Efficacy Outcome: Recurrent Ischemic Stroke
- Asundexian: 6.2%
- Placebo: 8.4%
- Cause-specific hazard ratio: 0.74 (95% CI 0.65-0.84; p<0.0001)
- 26% relative risk reduction
- Absolute risk reduction: ~2.2 percentage points
- Number needed to treat: ~45 patients for 2 years to prevent one recurrent ischemic stroke
Before we change practice, let's look at what the trial actually showed. This is a 26% relative reduction, which translates to preventing one stroke for every 53 patients treated over two years. That's clinically meaningful—not just statistically significant.
Secondary Efficacy Outcomes
- All strokes: 6.6% vs. 8.8% (HR 0.74, 95% CI 0.65-0.84)
- Disabling or fatal stroke: 2.1% vs. 3.0% (HR 0.69, 95% CI 0.55-0.87)—31% reduction in the outcomes that matter most
- CV death, MI, or stroke: 9.2% vs. 11.1% (HR 0.83, 95% CI 0.74-0.92)
- All-cause mortality, MI, or stroke: 10.5% vs. 12.3% (HR 0.85, 95% CI 0.77-0.95)
The consistency across secondary endpoints strengthens the primary finding. The 31% reduction in disabling or fatal stroke is particularly important—this is what patients and families care about.
Primary Safety Outcome: ISTH Major Bleeding
- Asundexian: 1.9%
- Placebo: 1.7%
- Cause-specific hazard ratio: 1.10 (95% CI 0.85-1.44; p=0.46)
- No significant increase in major bleeding
- No increase in intracranial hemorrhage
This is the critical finding. The effect size matters more than the p-value—and this effect size (HR 1.10, confidence interval crossing 1.0) means we have no evidence of excess bleeding. This is where FXIa inhibitors diverge from every other attempt to add antithrombotic intensity in non-cardioembolic stroke.
Subgroup Consistency
Benefit was consistent across age, sex, stroke versus TIA, stroke subtype (large-artery atherosclerosis, small-vessel disease, undetermined), NIHSS severity, use of thrombolysis, and antiplatelet strategy (single versus dual therapy).
When subgroups are this consistent, it suggests a robust, generalizable treatment effect.
What This Means for Practice
If these data hold up through regulatory review and FDA approval, asundexian would become the first new drug class for secondary stroke prevention in non-cardioembolic stroke in decades.
Who benefits: Patients with recent non-cardioembolic ischemic stroke or high-risk TIA who are already on antiplatelet therapy and need additional risk reduction. The trial enrolled patients with large-artery atherosclerosis, small-vessel disease, and cryptogenic stroke—all populations currently limited to antiplatelet therapy alone.
How to use it: Add 50 mg daily to standard antiplatelet therapy (aspirin with or without clopidogrel, depending on the clinical scenario and timing after stroke).
How long: OCEANIC-STROKE followed patients for 2 years. Whether indefinite therapy is needed or whether there's a high-risk window followed by de-escalation remains unclear. Duration of therapy will likely be guided by stroke subtype, ongoing atherosclerotic burden, and bleeding risk.
Who should NOT get it: Patients with atrial fibrillation requiring anticoagulation. OCEANIC-AF proved that asundexian cannot replace DOACs in this population. Patients with high bleeding risk would need individualized decision-making, but the OCEANIC-STROKE safety data are reassuring.
The Landscape — What's Coming Next
Asundexian isn't the only FXIa inhibitor in development.
Milvexian (Bristol Myers Squibb/Janssen): Two Phase 3 trials are ongoing. LIBREXIA-STROKE is testing milvexian in secondary stroke prevention—directly analogous to OCEANIC-STROKE. LIBREXIA-AF is testing milvexian in atrial fibrillation, despite the failure of OCEANIC-AF with asundexian. Topline data are expected in late 2026. The ACS trial (LIBREXIA-ACS) was discontinued.
Abelacimab (Anthos Therapeutics): A monoclonal antibody administered subcutaneously once monthly. AZALEA-TIMI 71 showed a 62% reduction in bleeding compared to rivaroxaban in atrial fibrillation, but efficacy data were less clear. LILAC-TIMI 76 (Phase 3) is taking a smarter approach—testing abelacimab versus placebo in DOAC-ineligible AF patients. This sidesteps the "is it as good as DOACs?" question and asks "is it better than nothing?" If you can't tolerate a DOAC due to bleeding risk, maybe near-complete FXIa suppression with a monthly injection is an option.
Asundexian positioning: Based on OCEANIC-STROKE, Bayer is expected to pursue FDA submission for secondary stroke prevention in non-cardioembolic stroke.
The Open Questions
Could higher doses work in atrial fibrillation? OCEANIC-AF failed at 50 mg versus apixaban. But what about 100 mg or 200 mg? Or is the mechanism simply wrong for AF thrombi regardless of dose? AZALEA-TIMI 71 with abelacimab (which achieves near-complete FXI suppression) showed similar stroke rates to rivaroxaban, suggesting that maybe near-total FXI inhibition could work in AF—but it hasn't been proven in a powered non-inferiority trial. The ongoing LIBREXIA-AF trial will add data here.
What's the right trial design for AF? LILAC-TIMI 76 is testing abelacimab versus placebo in DOAC-ineligible AF patients. This is a smarter question than trying to prove non-inferiority to apixaban. If FXIa inhibitors can reduce stroke in patients who can't take DOACs due to bleeding, that's a clinically meaningful niche.
Duration of therapy: How long should patients stay on FXIa inhibitors after stroke? OCEANIC-STROKE followed for 2 years. Should it be indefinite? Or is there a high-risk window (say, the first year) followed by transition back to antiplatelet monotherapy? Stroke recurrence risk isn't constant—it's highest in the first 90 days and declines over time. Whether FXIa inhibition should mirror that risk curve or continue indefinitely is unknown.
Which stroke subtypes benefit most? OCEANIC-STROKE enrolled 41% large-artery atherosclerosis, 29% undetermined, 22% small-vessel disease. Subgroup analyses showed consistent benefit, but larger atherosclerotic plaques (more thrombotic surface area, more FXIa amplification) might derive greater benefit. The PACIFIC-Stroke Phase 2 data hinted at this (HR 0.39 in patients with atherosclerotic plaque). Further subgroup analyses from OCEANIC-STROKE will clarify whether certain populations should be prioritized.
Combination strategies: Could FXIa inhibitor plus low-dose DOAC work in atrial fibrillation? Nobody has tested this. Could triple therapy (antiplatelet + FXIa inhibitor + low-dose DOAC) work in patients with both coronary disease and AF? These are mechanistically plausible but clinically unproven.
The Third Revolution
We can now divide the history of stroke antithrombotics into three eras:
- Era 1 (1950s-2000s): Antiplatelets for non-cardioembolic stroke, warfarin for atrial fibrillation. Aspirin, clopidogrel, aspirin-dipyridamole. Incremental gains, high bleeding risk with warfarin.
- Era 2 (2010s-2020s): DOACs replace warfarin for AF. Safer, more effective, easier to use. But nothing changes for non-cardioembolic stroke. We're still stuck with antiplatelets alone.
- Era 3 (2026 onward): FXIa inhibitors as adjunct therapy for non-cardioembolic stroke. A new mechanism that uncouples thrombosis from hemostasis, finally allowing us to push beyond the antiplatelet ceiling without unacceptable bleeding.
The road to Era 3 was bumpy. OCEANIC-AF failed spectacularly. LIBREXIA-ACS was stopped early. Phase 2 trials buried the signal in composite endpoints driven by covert infarcts that may or may not matter clinically.
But we learned where this drug class works. And OCEANIC-STROKE delivered: 26% stroke reduction, no excess bleeding, consistent benefit across subgroups, meaningful reduction in disabling and fatal stroke.
The numbers don't lie. This is clinically meaningful—not just statistically significant. After decades of stagnation in non-cardioembolic stroke prevention, we finally have a new tool.